
产品搜索
结构搜索
全站搜索
在线客服
-
诊断医药客服
当前位置: 专题聚焦
东北师大Nat. Chem.:吲哚的多样性分子编辑
本文转自x-mol
从结构简单的有机小分子出发,通过精确的外围编辑(peripheral editing)和骨架编辑(skeletal editing),构建结构多样性的分子骨架是现代有机合成的核心内容。近年来,分子编辑反应取得了巨大进展,但目前主要集中在C-H键官能化(外围编辑)上,很少用于潜在分子骨架的编辑。从简化逆合成的角度来看,单原子的“插入”和“删除”极具吸引力,例如Wolff、Favorskii和Bayer-Villiger重排等反应就能使碳、氮或氧原子插入到分子骨架当中或从分子骨架中删除,直接达到重塑分子框架的目的,与复杂的多步从头合成路线相比,更加符合“绿色化学”和“原子经济学”的理念。
吲哚是活性药物分子和天然产物中最常见的含氮杂环之一,被视为后期骨架编辑的理想底物。含氟片段的引入能够明显改善含吲哚或喹啉药物分子的生理活性(如已上市的抗疟疾药物- Mefloquine,图1c)。尽管关于吲哚分子编辑的研究已经取得了巨大的进展,但目前已报道的外围编辑和骨架编辑通常依赖于不同的反应策略和起始材料,仍具有一定的局限性。
东北师范大学毕锡和课题组长期致力于N-邻三氟甲基苯磺酰腙卡宾化学研究,其作为安全稳定的卡宾前体,实现了多种卡宾转移反应,解决了传统对甲基苯磺酰腙分解温度高、难以应用于低温反应的问题,开拓了磺酰腙卡宾化学研究新领域(Acc. Chem. Res. 2022, 55, 1763,点击阅读详细)。近日,该团队以氟烷基取代的N-邻三氟甲基苯磺酰腙作为卡宾前体,通过与过渡金属相互作用原位生成亲电氟烷基卡宾,同步实现了对吲哚分子骨架的多样性编辑反应。该研究从相同起始原料出发,通过对吲哚骨架或外围进行可调控的化学选择性编辑,实现了四种不同类型的反应:一碳插入反应、C3位偕二氟烯基化反应、双官能化反应以及C2,C3位环丙烷化反应,高效制备了各种含氟片段的N-杂环骨架(图1d)。这种分子编辑策略使用的起始原料简单易得,催化条件简单可控,为构建含氟片段的复杂分子铺平了道路。

图1. 研究背景和反应设计。图片来源:Nat. Chem.
吲哚是富电子芳烃,分子结构本身C3位和N1位均具有较高的反应活性,在与金属卡宾反应时可能会导致产生C2、C3位或N1位官能化产物和环丙烷化产物的混合物。为了实现N-无保护吲哚与氟烷基卡宾的发散性分子编辑,必须解决吲哚分子位点选择性和化学选择性问题。起初,作者主要集中在选择性骨架编辑反应研究上。反应条件优化实验表明,当吲哚为3.0倍量时,在80 ºC下,TpBr3Ag(thf)能够催化反应以96%的产率得到插碳产物3。而将吲哚降低至1.0 倍量,N-邻三氟甲基苯磺酰腙提高至3.0倍量,以1,2-二氯乙烷(DCE)为溶剂时,在60 ºC下即可实现双官能化反应的高效转化。碱的种类对反应亦有影响,当使用N,N-二异丙基乙胺(DIPEA)时,可促进脱氟反应的发生,成功实现了氟烷基卡宾对N-未保护吲哚的C3位偕二氟烯基化反应(表1,条目6),这在先前报道的工作中是从未被实现的。
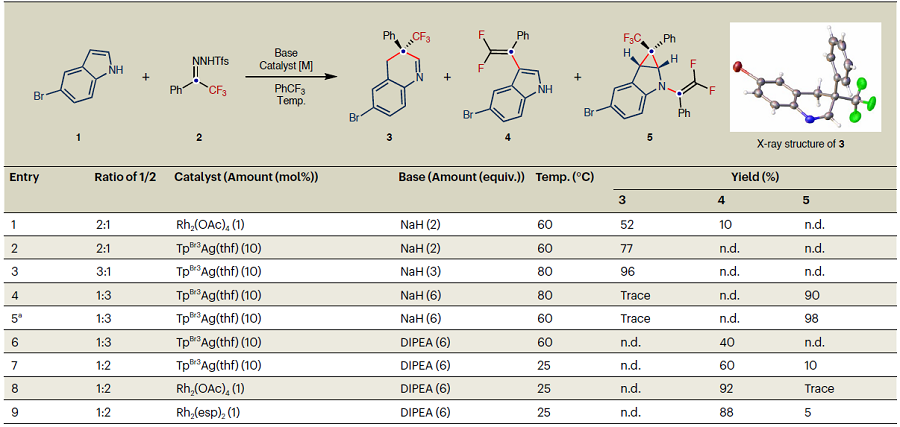
表1. 反应条件优化。表格来源:Nat. Chem.
银催化的氟烷基卡宾对吲哚的一碳插入反应具有宽泛的底物适用范围和良好的官能团耐受性。各种拉电子基、给电子基以及杂芳环取代N-邻三氟甲基苯磺酰基腙和吲哚均可以良好至优异的产率得到一碳插入产物。其中,全氟烷基链的引入以及4-氮杂吲哚和6-氮杂吲哚的成功转化,将对新药研发起到促进作用。为了验证该方法的高效性和实用性,作者进行了一锅两步反应、克级规模实验以及系列衍生化实验,成功得到了四氢喹啉、3,4-二氢喹啉-2(1H)-酮等产物。
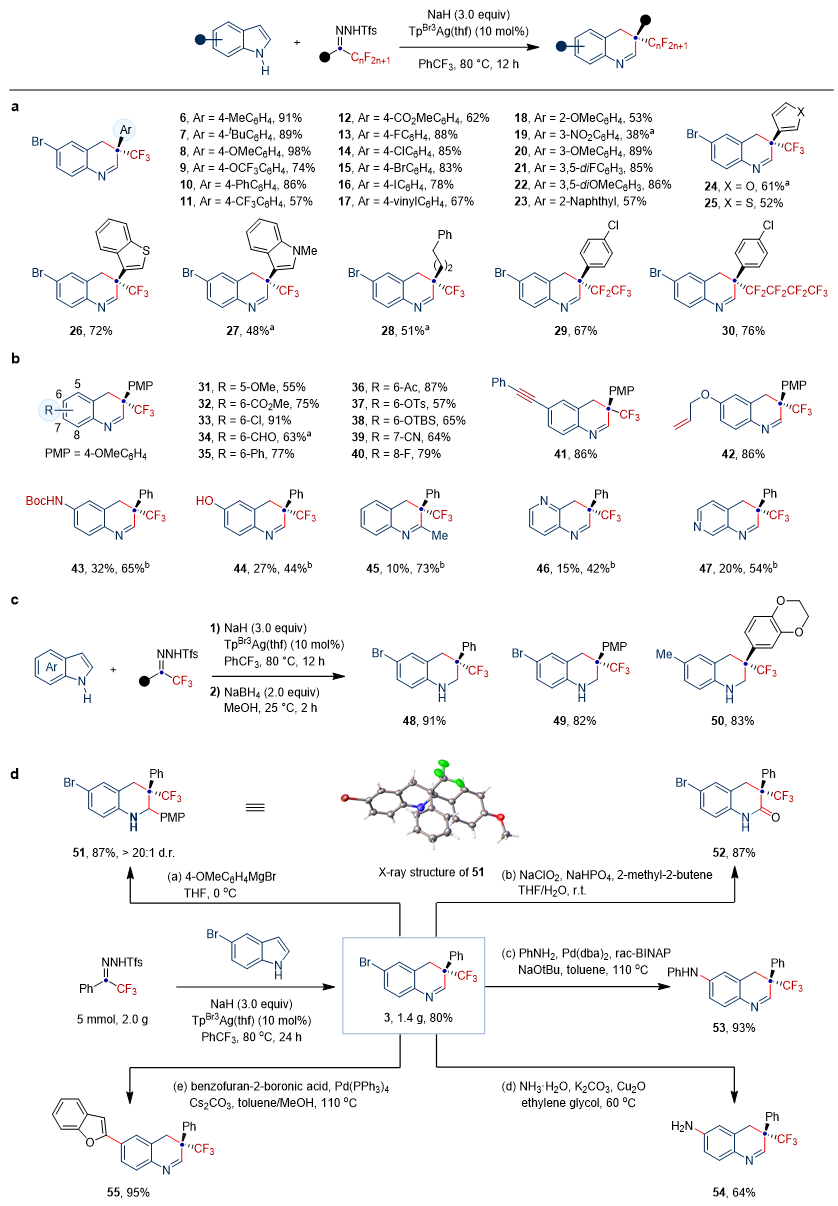
图2. 氟烷基卡宾对吲哚骨架碳插入反应研究。图片来源:Nat. Chem.
紧接着,作者对吲哚C3位偕二氟烯基化反应的底物范围进行了考察。如图3a所示,无论是N-未保护吲哚还是N-保护吲哚,一系列具有各种功能手柄的N-邻三氟甲基苯磺酰基腙均能够实现反应的高效转化。吡咯C2位偕二氟烯基化产物的生成,进一步展示出了该方法的普适性,这在先前报道的文献中是难以实现的。此外,双官能化反应也显示出了较好的官能团耐受性。对于N-保护吲哚而言,除了C3位的协二氟烯基化反应,同样可以实现C2,C3位的去芳构化环丙烷化反应。如图3c所示,无论是变换吲哚上的取代基,还是更替N-邻三氟甲基苯磺酰基腙上的取代基,甚至是吲哚N原子上的保护基,如苄基、芳基、特戊酰基(Piv)、氨基甲酰基(CONMe2)、叔丁基二甲基甲硅烷基(TBS)等官能化基团均可以兼容到该反应体系当中。
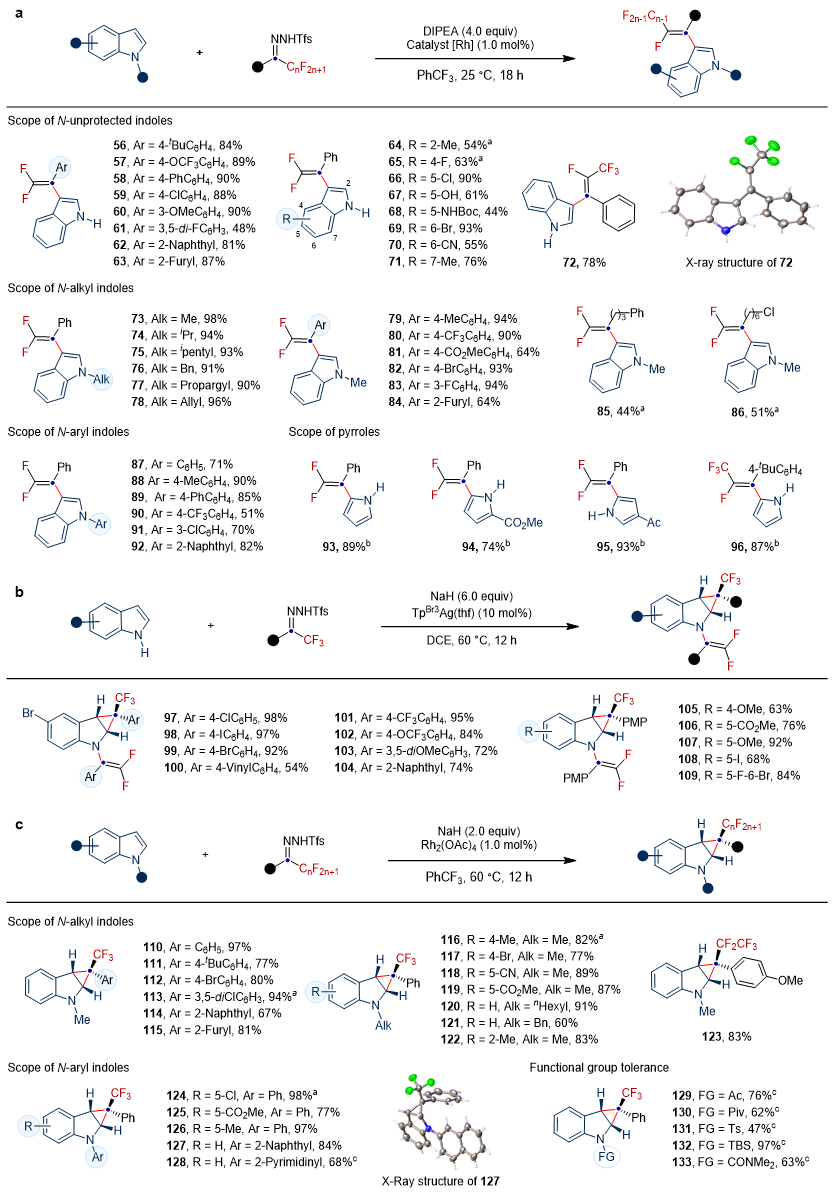
图3. 氟烷基卡宾对吲哚的外围编辑反应研究。图片来源:Nat. Chem.
偕二氟烯烃可作为反应活性手柄,作者通过其与苯甲酰肼的环化反应,制备合成了2,5-二取代的1,3,4-恶二唑(134),展示了偕二氟烯基化产物的应用价值。鉴于吲哚的骨架编辑和外围编辑可能促进新药的研发以及三氟甲基和偕二氟烯基在药物化学中的重要性,作者对两种从新热带植物中分离出来的天然产物verticillatine B和raputimonoindole B进行了多样性分子编辑反应,在不同的反应条件下,分别实现了两种天然产物的一碳插入反应、C3位偕二氟烯基化反应以及双官能化反应,为吲哚或喹啉类新药的开发提供了依据。
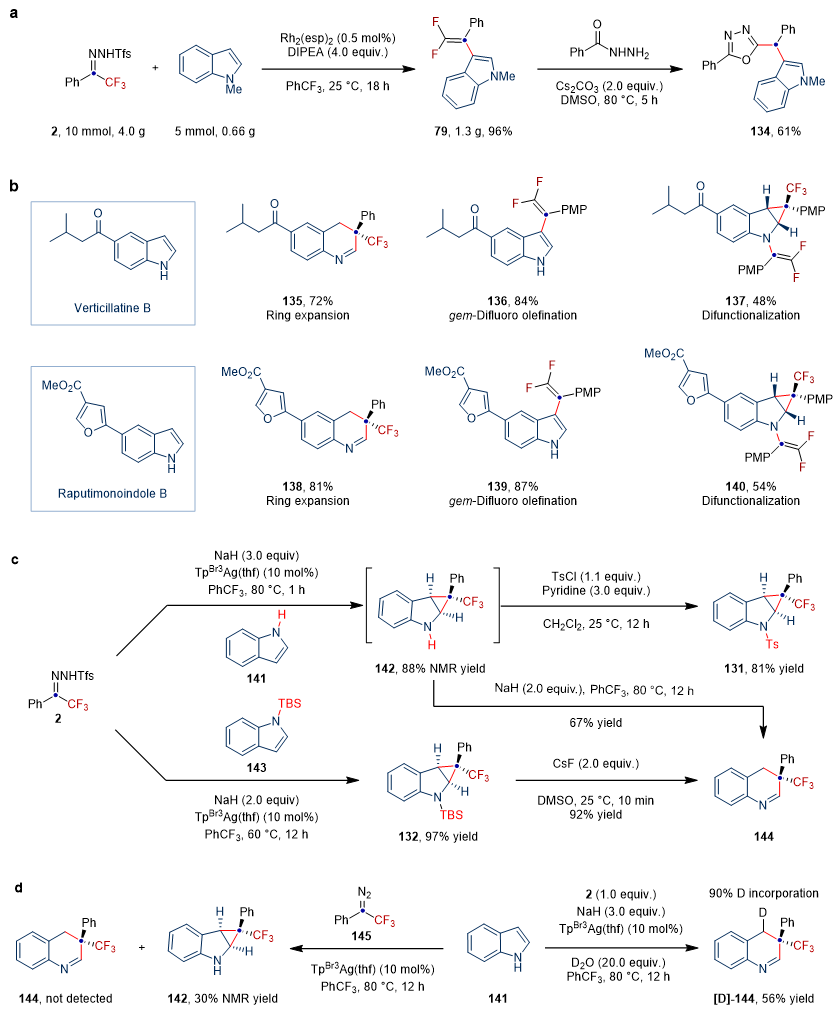
图4. 衍生化实验、天然产物后修饰及机理验证试验。图片来源:Nat. Chem.
为了深入研究反应机制,作者进行了系列对照实验(图4c和4d)。首先,经过核磁氢谱分析,底物2和141在短时间内(1 h)可以得到环丙烷化产物142(由于该环丙烷化产物142稳定性较差,不足以通过柱层析色谱法分离),同时该环丙烷化产物可被对甲苯磺酰氯捕获得到环丙烷产物131。相反,N-TBS保护的环丙烷产物132,可在室温下脱除TBS基团进而转化为插碳产物144(图4c)。这些结果均表明,插碳反应过程可能经历环丙烷中间体。为了验证NaH在开环过程中的作用,作者以苯基三氟甲基重氮甲烷为卡宾前体,反应体系中不加入NaH时,未监测到碳插入产物生成,这表明NaH在开环反应过程中是必不可少的。此外,氘代实验结果表明,反应溶剂中微量的水协助参与了质子化过程(图4d)。
为了进一步探究反应机制,作者进行了理论计算研究(图5)。Fukui函数分析表明,吲哚C3位相较于N1的亲核性位更强(图5b),亲电金属卡宾优先进攻C3位,这与实验结果是相一致的。当使用Rh2(OAc)4-DIPEA催化体系时,DIPEAH+能够促进β-F消除过程优先发生,生成C3位偕二氟烯基化产物146。该过程中DIPEA和F原子之间的氢键相互作用对降低吉布斯自由能起到了至关重要的作用(图5c)。吲哚对卡宾IntI-Rh的亲核进攻为Rh催化偕二氟烯基化的决速步骤(ΔG≠ = 12.5 kcal mol–1)。而TpBr3Ag–NaH催化剂体系中不存在这种氢键效应,优先发生C2,C3位环丙烷化过程(图5d)。当吲哚过量时,环丙烷中间体IntIII经过NaH拔氢,可逆开环以及水协助质子化过程,得到一碳插入产物144。当N-邻三氟甲基苯磺酰腙过量时,形成动力学上有利的叶立德中间体IntIV1,再经历β-F消除过程得到双官能化产物145,这与实验结果均一致。
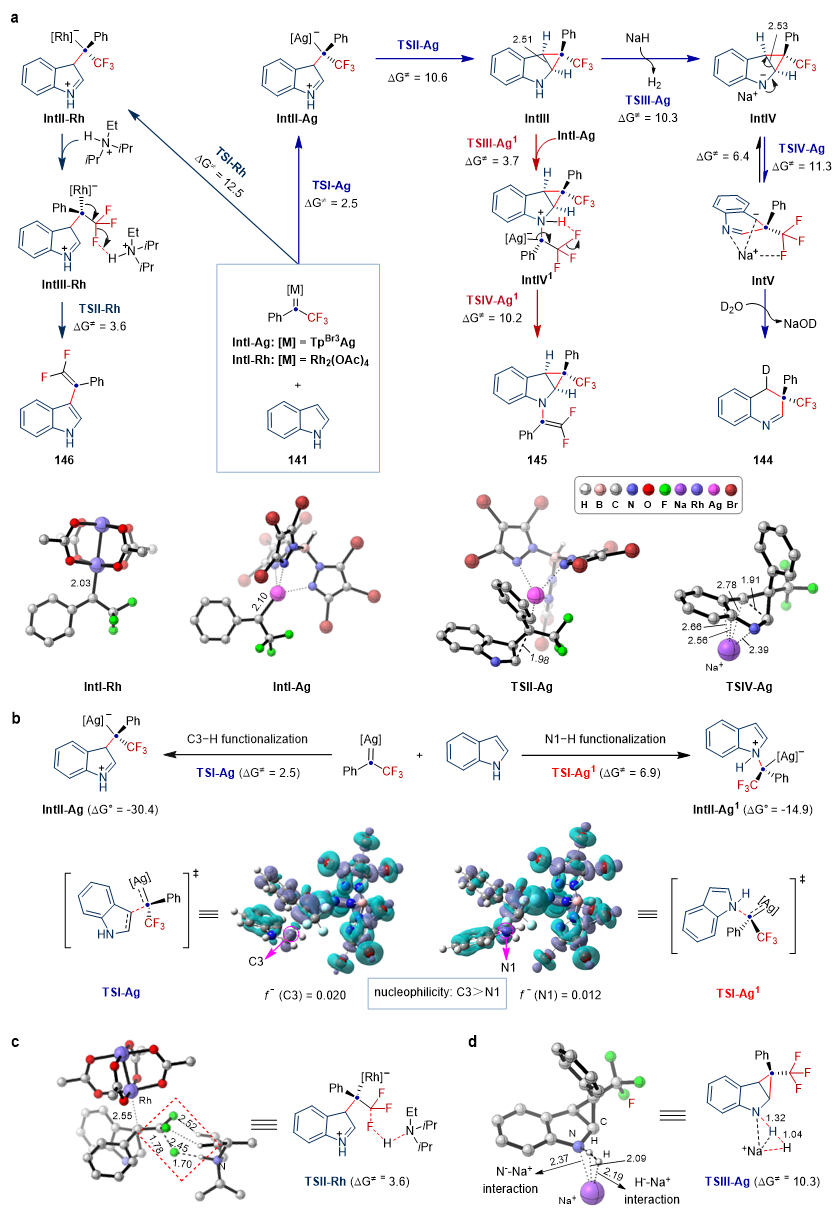
图5. 反应机理研究。图片来源:Nat. Chem.
为了探索催化剂对反应路径的催化机制,作者分别比较了Rh和Ag催化生成144、145和146的反应路径。结果表明,Rh和Ag催化剂均可以催化反应体系生成三类产物。C3位偕二氟烯基化过程和C2,C3位环丙烷化过程取决于碱的种类。当NaH为碱时,Rh和Ag均可催化生成产物144和145,反应选择性取决于N-邻三氟甲基苯磺酰腙和吲哚的比例。当碱为DIPEA时,Rh催化时形成产物146和145分决速步骤差(ΔΔG≠ = 7.4 kcal mol–1),比Ag催化体系(ΔΔG≠ = 2.3 kcal mol–1)大,这决定了在C3位偕二氟烯基化反应中,Rh催化比Ag催化具有更高的选择性。这与实验观察到的结果保持一致。
小结
毕锡和教授课题组以氟烷基-N-邻三氟甲基苯磺酰腙作为卡宾前体,在银或铑的催化下,实现对吲哚分子可控的单原子骨架编辑和外围编辑反应。该策略中,金属催化剂和碱的组合是实现可控分子编辑的关键。该策略所使用的卡宾前体稳定易得、操作安全,反应条件温和、官能团耐受性好、产物结构多样性高,高效合成了多种含氟片段的N-杂芳烃骨架。
这一研究成果近期发表在Nature Chemistry,东北师范大学刘兆洪副教授和毕锡和教授为通讯作者。博士研究生刘少鹏、杨勇、宋清敏为共同第一作者,硕士研究生王占静、陆颖参与实验合成。该研究工作得到了国家自然科学基金重点项目(22331004,21871043,21961130376)、面上项目(22371035)等大力支持。











